Hello, my name’s Elizabeth Blackburn. I’m in the Department of Biochemistry and Biophysics at the University of California, San Francisco. And in this set of lectures, I’m going to talk about telomeres and telomerase. And I’ll get to their implications for human health and disease. The first part of this series of three lectures is going to introduce you to the roles of telomeres and telomerase. So, let’s begin by focusing in on what goes on at the very heart of a cell. Now if you looked at a cell which is just about to divide, you looked into the microscope and you stained the chromosomes, this is what you would see.
Those blue, double sausage-like objects that you can see all over this microscope slide are the human chromosomes, and the DNA has just duplicated, which is why they look double. Now if you look closely, you can see red spots, two red spots at the ends of every double chromosome pair. And these red spots are a molecular probe that’s lighting up the telomeric DNA that’s found in common at all of the chromosome ends. And I’m going to tell you a lot about that telomeric DNA. So why are telomeres important? Their role is to cap off the ends of chromosomes.
So that’s a simple concept, but we can dive down into it further and think a little bit more about what that actually means. So when we think of the end of the DNA, there can be two kinds: There can be the natural end of the DNA, such as the ones we see here, the ends of the chromosomal DNA; and there can also be DNA breaks. Now, the job of a cell is to seal up any DNA breaks that happen by accident. And so a major part of this capping function, which is one of the aspects of telomere functions, is to prevent the telomeres from undergoing those very DNA transactions of very DNA reactions that are undergone by a broken DNA end. So if you have a break in the DNA, it can be sutured together by, for example, recombination or just simply the two broken ends can be ligated right back together by end-to-end fusions.
Also, such DNA breaks are subject to degradation. Now the telomere protects, it caps, the end of the chromosome and protects against all of these kinds of things that would normally happen to a broken DNA end. How does it do that? The DNA sequences that you find at the ends of chromosomes, repeated over and over, are fairly similar in nature to each other. They’re relatively simple sequences, and they’re too simple to code for any proteins. They’re not genes in the sense of coding for any proteins or RNAs. In humans for example, this repeated sequence is found up to a few thousand times, tandemly repeated over and over at the ends of the chromosomes. Another feature of the chromosomal DNAs is that, of course, unlike most of the DNA of a chromosome, which is duplex DNA, double-stranded DNA, the very end is single-stranded.
And in fact the DNA strand is oriented 5′ to 3′, going toward the end of the chromosome. And that turns out to be important. So, we have now the telomere structure to begin with: We have again, if we blow up the end of a chromosome, we have these highly repeated sequences made up of a G- rich sequence that’s the repeat unit repeated over and over again. As I said, it doesn’t encode any protein sequences, but each of these repeated sequences is like a little attractive magnet for specific proteins that bind sequence specifically to the telomeric DNA. They bind to the telomeric repeats for the double-stranded portion, and some of them bind the single-stranded portion, and that’s actually the G-rich strand that’s forming the overhanging end here. These together make some form of higher order architecture we don’t understand. We understand a lot about the protein-DNA interactions, some of the molecular details of that. Some of the details of the protein-protein interactions in this complex, but we don’t really understand the higher order structure, so that’s still a challenge in the field.
So I’ve just shown you a functional telomere, but if telomeres cease to function… and we use the term “telomere dysfunction” to just describe that general state of the telomere that is not carrying out those capping functions and other functions that I will get to… there are a couple of different ways this can happen. The first is if the tract of telomeric repeat is simply too short, there’s just not enough of the length of the repeats to form a nice, long array that can form this higher order structure that’s necessary. This kind of dysfunction caused by the shortening of the telomere, that can happen naturally, and it does, and we’ll get back to that in a moment, because this is going to be an important part of these lectures, and in fact it’ll be really the focus of the third lecture in this series.
The other way that telomeres can become dysfunctional: If for one reason or another, experimentally induced, most commonly, one of these proteins cannot bind correctly to the telomeric DNA; if its binding is disrupted through some molecular intervention or other… in both cases, cells sense and respond to this state of telomere dysfunction. Now indeed, cells have a lot of very strict regulatory reactions to the lack of proper telomeric DNA. And the consequences for the cell is that, usually this state of the cell’s telomere dysfunction, through one reason or another, will mean that the cell will cease to divide. So this limits cell renewal capability if this happens to one or more of its telomeres in the cell. If by chance the cell does continue to multiply, now those telomeres become subject to the very kinds of fusions (the DNA- joining events that I told you telomeres shouldn’t allow to happen), and that can lead to genomic instability, because the end-to-end joining of telomeres to themselves (other telomeres, that is) or to broken DNAs, that can cause the chromosomes, which fuse to each other, to tear themselves apart as the cells divide, leading to genomic instability.
So clearly telomere function is very important for cells. And in fact, one of the consequences of genomic instability in human cells is that the cells can become cancerous. I’m just going to show you a picture. If you look under a microscope of some cells in which we’ve disrupted one of the telomeric proteins, what you can see is… remember I told you the blue, double things are the chromosomal DNAs… and look, here’s a chromosome here in which there’s been a telomere fusion.
So here’s the two telomeres at the end, here’s the other two all the way here, but there are fused telomeres here. So this is now a chromosome that has two centromeres, it’s got a centromere here, it’s a got a centromere here, and if those two centromeres try to pull apart in a cell that is dividing, the chromosome will get ripped apart. Here’s another example of such an end-to- end fused chromosome. This kind of change can happen if you disrupt the telomeric integrity by, for example, disrupting the binding of proteins. The other kind of function, it’s related, but we can distinguish it, the other kind of function of telomeres is that they have to allow for the complete replication of the telomeric DNA.
So what’s the issue here? Well, the mechanism of DNA replication, the machinery of DNA replication, has a particular quirk to it. It’s very good at faithfully copying almost all the way along the length of the chromosomal DNA (or any linear DNA), but the make-up of the DNA replication machinery is such that is cannot copy the very, very end of the linear DNA, such as a eukaryotic chromosomal DNA. Now the predicted and observed consequence of that inability is that, each time the DNA replicates, which is has to do as the cell divides, and then the cell divides, the daughter DNAs are predicted to become shorter and shorter and shorter. And this is just a simple consequence of the nature of the DNA replication machinery that is otherwise so good at replicating all the way along all the length of the chromosomal DNA. But the very ends cannot be completely replicated without some form of compensatory mechanism. So what’s the consequence of this loss? Well, obviously, something has to compensate in the long run, otherwise, we wouldn’t be here.
But even on a shorter timeframe, as cells divide and divide, the DNA gets shorter and shorter, one might predict that there would come a point when there wouldn’t be enough of something or other at the end that then the chromosomes would no longer be able to support cell division, and the cells might eventually undergo what’s called senescence. And indeed James Watson in 1972, just considering the mechanism of DNA replication, proposed this constant shortening problem, and Olovnikov around the same time proposed that in fact perhaps such loss of terminal DNA, without knowing at that stage what the molecular nature of the terminal DNA was, Olovnikov proposed that perhaps such gradual loss could be something that underlies the eventual senescence of cells that is seen sometimes when, for example, human cells are grown in culture.
This was a prescient idea because, in fact, this indeed has been found to be one of the causes of why human cells cannot replicate themselves, the cells cannot proliferate, indefinitely in culture. So, shortening of telomeric DNA is something that will be problematic for cells. How is this problem solved? Well, it’s solved by an enzyme called telomerase. So I’d like to introduce you to telomerase now, and how it was found. Telomerase was sought because there was a set of accumulating observations on telomeric DNA in cells, the telomeric DNA as it was in cells in vivo, that couldn’t be readily explained by what was currently known about DNA replication or DNA recombination or other kinds of DNA reactions at the time, which was the late 1970s, early 1980s. And let me give you some examples of such puzzling observations. Well, the first one was that in the ciliated protozoan Tetrahymena, which has a lot of very small mini-chromosomes and is therefore amenable to molecular analyses of its telomeric DNA by direct methods, the telomeric repeat sequences (remember I told you about the repeated sequences repeated over and over at the ends of chromosomes)…
The repeat sequence in this organism, G4T2 repeats, were heterogeneous in their number, in different molecules in a population of otherwise homogeneous cells. And heterogeneous in this setting here is meaning, these were different in number, so some copies of the mini-chromosome would have twenty repeats on the end, some would have 50-some, 49-some, 82-some, 53… they all had different numbers of repeats in this sort of more-or-less normal distribution. So that was very surprising, because if you look across at the internal region of a DNA, such as a chromosomal DNA, if you look at one cell compared with another in a population of cells from, say, one organism, it should always be an identical sequence. So here were different numbers of repeats at the ends of different molecules in a population of cells that should have otherwise been homogeneous, and were homogeneous in their internal regions of the chromosome. Now, a second kind of observation was a somewhat more complicated one, and it means that I have to just take a moment to tell you about the lifecycle of a particular group of ciliated protozoans, which the species Tetrahymena in which these G4T2 repeat tracts were found to be the telomeric tracts.
The Tetrahymena cells go through a lifecycle stage where they have a somatic nucleus which undergoes developmentally controlled fragmentation. And fascinatingly, telomeric DNA sequences were added directly to those freshly formed DNA ends, making from longer chromosomes a series of shorter mini-chromosomes. How did that telomeric DNA get added to the ends? It wasn’t clear. The third observation came from observing the cells of an organism which causes sleeping sickness, a single-celled parasitic organism called a trypanosome.
And these were being propagated in the laboratory setting, and what was found was that the telomeric DNA restriction fragments, the end fragments of the chromosomes in these organisms, were gradually getting longer and longer and longer. And this didn’t look like, say, recombination, and certainly was not expected for normal, as one thought about it, DNA replication. The fourth observation was again something that came out an experiment that I’ll have to explain. Now one could put circular plasmids into yeast cells, and those plasmids are linearized, essentially they’re unstable, they get gobbled up or, rarely, will recombine into the chromosomes and thus be preserved.
But what was found was that if one simply grafted onto the ends of such a linearized and therefore normally very unstable yeast plasmid, if one grafted onto its ends Tetrahymena telomeric DNA fragments (the telomeres of Tetrahymena mini- chromosomes) and introduced those into cells… so the grafting was done in vitro with enzymes and the purified DNA molecules, and then the resulting hybrid of the yeast linearized plasmid and the telomeres added onto its end, that was introduced into the yeast cells, those were maintained in yeast cells as linear mini-chromosomes, and yeast telomeric DNA repeat were grafted on somehow inside the yeast cells, to the ends of the Tetrahymena telomeric repeats.
And that didn’t look like a reaction one would expect from standard known models of DNA replication or recombination. All of these suggested that perhaps there was some capability of cells to add telomeres. And that idea, in a conceptual way, was given some force by an observation made by the noted geneticist, Barbara McClintock, who worked with maize (corn), and she noted that a particular maize mutant stock lost the capacity to do something that is normally found in normal, wild-type maize.
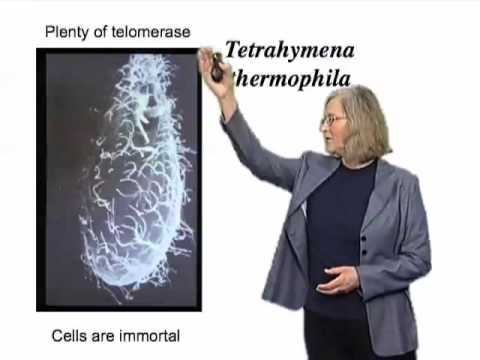
In such wild-type maize, if a chromosome is broken by, for example, exo-radiation or some mechanical rupture at a particular stage in development, then that broken end can be, as she described it, “healed.” It becomes a normal, stable telomere. Nobody knew the molecular basis of that. And she found a mutant that had lost that capacity. And when you see a mutant where something is changed, you have a feeling that that’s reflective of a cellular process that can take place, and that it’s not just by chance that this healing event was taking place. All of which, then, focused in on the question of: Was there a new enzyme that worked in cells that could extend telomeric DNA? So we sought such an enzyme in the early to mid-1980s. And we used for that purpose the single-celled, ciliated protozoan Tetrahymena thermophila, shown in this scanning electron micrograph here. And you can see the cilia are on the cell surface. This experimental system was chosen because the organism contains large numbers of very short mini-chromosome, therefore, large numbers of telomeres, and therefore, one would reason, perhaps if there were such an enzyme that existed that could add telomeric DNA to the end of chromosomes or to preexisting telomeres, then this would be a potentially good source for it because there’s a lot of telomeres in this organism.
And indeed that proved to be the case. And Carol Greider, my then-graduate student, joined the lab in 1984, and here’s a picture of Carol, freshly from visiting Southern California, and looking at an autoradiogram shown with this x-ray film here in the lab. And we together found this enzyme telomerase. Now let me show you briefly what we did. We took a mimic of that overhanging G-rich strand DNA that’s normally found at the ends of chromosomes, in the form of an oligonucleotide, and here it is shown here. And I’ve just colored the bases different colors so that we can see them easily. And here’s its 3′ hydroxyl end. We mixed it with an extract of Tetrahymena cells, and this worked particularly well at the stage in development that I mentioned to you earlier… that stage at which telomeric DNA is added to freshly broken ends of chromosomes. We reasoned that that would be a particularly good time to find such an activity, thinking it might be likely to be induced or present in perhaps larger-than-normal amounts, because this would be a time when there’d be a demand for such an enzyme.
All of this was hypothetical at the time, but by putting in an appropriate mix of things that are often added to make polymerases happy (such as magnesium ion), and just by using two nucleoside triphosphate precursors, dGTP and TTP, we were able to find that the telomeric DNA indeed was added to the end of such an oligonucleotide. In fact, in the test tube, a large number of repeats could be added. So we get a lot of repeats added, through eventually something limits the addition. So this is what telomerase did. Now how is it doing that? It was adding a given sequence. The first clue came from the observation… and here I’m just showing you a nice picture of the products run on a DNA sequencing gel, just to give you feel for what it looks like. We’ll just look at one of these groups, so here are different time points, increasing number of minutes after incubating this input primer with dGTP and TTP. The dGTP was radiolabeled, so we’re just looking at an autoradiogram.
Every time we see a labeled band, this is a product that’s been added, so the input would run here. If you added one, two, three, four nucleotides, you could see you’d get longer and longer bands. And what you can see is that there is kind of a striped pattern. Every six nucleotides, there was a pause in the addition, and so you could see a pattern of six nucleotides being added, and you could see more and more repeats, as shown by the bands getting higher and higher and higher, were being added with time. Now, certain clues came. Not any nucleotide could work. So, two examples of the telomeric DNA, where we’ve got a permutation ending with four Gs here, or a permutation ending with two Ts here. These were each competent for addition by this activity. However, the complementary strand, and again, I’ve just colored the bases distinctively and given you two examples… the complementary strand oligonucleotides were not competent for such addition.
Another interesting clue came when we looked in more detail at different oligonucleotides that could act as primers. Now, we had found, as I said to you, that when you put Tetrahymena telomeric repeats into yeast cells, live cells, then yeast repeats were added somehow, it was unknown then, to the Tetrahymena telomeres. And the yeast repeats have a different sequence, and I’ve shown you a little segment of it. It’s T, G, sometimes one G, sometimes two, sometimes three Gs. So we wondered if the converse would be the case; if we added a yeast primer to a Tetrahymena extract, now in vitro, would we see Tetrahymena sequences added to the yeast telomeric oligonucleotide. And indeed, we did see such addition. And again, large numbers of repeats could be added to such a primer quite efficiently.
Now we also noticed another interesting thing about the reaction. When we had a primer that ended with four Gs, what we found was that first two Ts were added, and then four Gs, two Ts, and so on. When we had a primer that ended with three Gs, first a G was added, and then the two Ts, in other words completing this run of four Gs.
And I haven’t shown it, but if you had a primer that ended in two Gs, then GG, and then TT. So this suggested that perhaps something was aligning this very first portion, the portion of the reaction that’s taking place, adding to the very 3′ end of the primer, that perhaps something was aligning this, in the enzyme somehow. Because in other words, if you lined it up like this, then everything was lined up.
What could be doing this? So was there something that aligned the product-forming part of this enzyme? Remember this was very mysterious at the time. So in fact we looked and found that, indeed, as I said, you had four Gs, and you added two Ts. If you had three Gs, one G was added. Two Gs, and you’d have two Gs added, and then the two Ts.
And if you had one G, then it would be three Gs, and then the two Ts. So in fact, there was something that was aligning how the next repeats were added, dependent on the 3′ end of the primer. The result of all of this kind of analysis led us to the idea that there was in fact a template within the telomerase, and to our surprise, this template turned out to be made of RNA, an RNA that is actually built in to the telomerase complex. And this template is a short portion of this RNA, which otherwise has a lot of other structure which is built into the enzyme particle, which contains also a protein, which is called TERT. So what telomerase does is it takes that single- stranded G-rich strand, it aligns the 3′ end nucleotides by Watson-Crick base pairing onto the template sequence, so here’s the example where we have two Gs at the end of the primer. It aligns it on this part of the sequence, and then it polymerizes, one at a time, each of these nucleotides onto the DNA end, extending the DNA end by copying this template.
And so now, you end up with longer DNA. So telomerase is a unique polymerase, it’s a reverse transcriptase by the definition, that is, copies RNA into DNA. It’s unique because the RNA component is actually intrinsically built into the telomerase ribonucleoprotein particle. It has to be built in for that templating to take place. And indeed the enzyme is truly a ribonucleoprotein enzyme, we believe. And unlike, for example, the reverse transcriptases that copy, say, the HIV viral genome, which is a genome thousands of nucleotides long, a very complicated sequence which encodes proteins, this enzyme copies very short sequences over and over again.
And it does it by this process of aligning the DNA end on the template, and so just to complete the thought here, here’s the template. If we polymerize all these together onto the DNA end, now you’ll have a new DNA end that ends with TTG, and that will realign in the next round back in this AAC, for another round of synthesis, and this can go on in the test tube for many repeats. I’ve told you about some of the enzymatic properties of telomerase, but what good is telomerase for cells? Well, the answer came by manipulating telomerase in Tetrahymena, the organism in which it was first discovered.
So if you remember, telomerase is adding telomeric DNA to the ends of chromosomes, and so the question was: What happened if it couldn’t do that? So we looked in Tetrahymena, which, as I said, was a good source of telomerase, and another feature of these organisms that I didn’t tell you was that, if you grow them in culture in the laboratory, they’re effectively immortal, so long as you keep them fed and under good conditions, they can just propagate and propagate and propagate, seemingly forever. And they have plenty of telomerase. We manipulated the telomerase RNA. Now, we made some changes in the RNA, and I won’t go into the details of it, but the effect of such small changes in the RNA I’ve shown diagrammatically here. The telomeric DNA repeat sequences, which as I said consist of multiple repeats at the ends of chromosomes normally, as the cells went through cell divisions, the telomeres progressively got shorter and shorter, and after about 20 or 25 cell divisions, the cells ceased to divide.
So in other words, when we inactivated telomerase, over the succeeding cell divisions the telomeres progressively shortened, so in effect when the cells ceased dividing, they’d become mortal. From being immortal, they’d become mortal. And all we had done was to inactivate telomerase by this small change, so like a little stiletto stuck at the heart of telomerase, we killed the enzyme very surgically, and we were able to make immortal cells become mortal. So, the conclusion is that the telomerase maintains the ends of chromosomes. Telomeres are replenished by telomerase as they keep dividing. And in fact, that continuing replenishment, even in the face of the continuing shortening processes that take place, can compensate for those shortening processes and allow the cells to keep on dividing. So that’s what telomerase does for cells. And that’s the important message: Telomeres are replenished by telomerase. Now, let’s go into a more detailed experiment in yeast cells.
If we look at a culture of yeast cells and we plate them out on a plate in a laboratory… all those little white spots are cells that have grown up from individual cells distributed on the plate, they’ve been distributed on kind of a V-shape pattern here, a triangular pattern. So, the cells are growing well normally. Now if we delete one of the genes for telomerase, either the RNA structural gene or the protein structural gene for the core of the enzyme, now just as I described for you in Tetrahymena, the cells divide and divide, and the telomeres progressively get shorter and shorter, until they get to a point where most of the cells completely cease to divide.
And so now they’re no longer capable of producing a rich growth of colonies on such a plate. See, there’s very little growth here. And we call that senescence. So taking away telomerase is a bit of a delay as the cells go through about 50 or so cell generations, 50 to 80, and then the cells’ telomeres get too short, and they undergo senescence. We’ve scrutinized more closely what happens to cells at this point. Interestingly, a few cells do survive, and in fact, those cells eventually become quite capable of producing good growth on a plate, but they’re different.
These cells now have very heterogeneous and long telomeres. I’ll show you these in a more graphical form in a moment. A gene required for this to happen, for such “survivors,” as we call them, to appear, is the gene RAD52. For at least one of these pathways, the RAD50 gene is also required. What are RAD52 and RAD50? These are in the homologous recombination pathway. They’re needed for recombination and therefore, to get survivors required recombination. Similar phenomena have been seen in mammalian cells. They’ve been less well characterized genetically, but similar survivor cells have been seen, and they’re called ALT cells in mammals, for “alternative lengthening of telomeres”… ALT cells. So, without telomerase, most cells go through senescence, but rare cells can survive. So, that is another way that chromosomes can survive, but interestingly enough, ALT is not normally seen in most normal cells. Now we wondered why, because on the face of it, it looks as if these cells are growing perfectly well. Although if you look more closely, you will see that some of the cells are not living well, but the great majority can survive with these very long telomeres, that they can keep recombining, and keep the population up that way.
We asked what’s different. So first of all, we quantified very carefully the cell growth, so this is just one way of showing it. The colony- forming units, which is a measure of viability, as the cells went through the progression of shortening telomeres, getting to this point when they get to senescence, and then those rare survivors now overgrew, and now you can see them taking over and becoming the population. So they’re growing again quite well. And we looked at the gene expression profile in such cells. First of all we looked at the telomeres, just to make sure that the telomeres were doing what I showed you diagrammatically before, yes they are, so here’s the telomeres. And this is what’s called a Southern blot, where we’re probing with a telomeric DNA sequence. And the most important are these bands down here, that’s the easiest to look at initially, because this is a lot of the telomeres. You can see they’re of a certain length, and as they get shorter they run faster and faster in the gel, so you can see they’re getting shorter and shorter with these successive days of passaging of cell divisions without any telomerase present.
Now, there are very few cells here actually, but we loaded enough DNA so you can see what little telomeric DNA there is. And then the cells start growing well again, and what you see is now the telomeres, the pattern has changed, they’re very heterogeneous, and there’s much longer telomeres, you can see they’re longer than these ones here. So that’s the telomeric profile. Now, let’s look at the gene expression profile. This is what’s called a microarray experiment, and in this experiment, one compares the pattern of gene expression by looking at the messenger RNA levels for all of the genes in the genome. And one asks, does a particular gene, compared with a reference, which is right at the beginning, when everything is growing well, does a particular gene’s level of expression become higher or lower. If it becomes lower, it gets greener and greener. And so each of these going across is a time point, each line in the column represent a single gene, and each of these horizontal areas represents the day at which the RNA was taken out of the cells and analyzed in this way.
And so what we find, and it’s all compressed and you just need to look at the pattern, was that about 650 genes changed their expression, and they got either lower levels of expression (and we’ll talk about this in more detail, and that’s shown in the green) or higher levels of expression (and that shows up as red). Now right away what you can see is that the maximum changes occurred right when the cells were approaching and into senescence. Six- hundred and fifty genes is something like 10% of the genes in the genome, which is about 6000 to 7000 genes. So about 10% of the genome shows a change in expression, particularly at senescence, but also, as I’ll show you, some genes remain changed even when the cells are growing well, maintaining their telomeres through this recombination mechanism. So one can analyze all the patterns of genes whose expression has changed and compare them with known patterns of gene expression changes that have been looked at in a variety of experimental settings by the many people in the yeast genomics and gene expression community, who’ve looked at gene expression changes.
So one can compare what we see here with what’s been seen in other experiments done by many other groups looking at many other conditions of changes to the cells in yeast. So what we found was that in fact, this pattern we observed was unique, and we gave it a name: the telomerase deletion response, the TDR. Now we found a set of genes uniquely upregulated in response to the deletion of telomerase RNA, and we called that the “telomerase deletion signature.” It was a particular group that behaved as a group only when you deleted the telomerase RNA, so that would be a group of those that showed up as red, because they were upregulated. And that hadn’t been seen by any other manipulation.
So the cells indeed were sensing that they were losing telomerase, and they responded in a particular way, by changing their physiology, which we read out as a change in gene expression profile. Right at the stage of senescence, when I showed you that there were a lot of genes that particularly were turned up or down in their activity, there was a particular known DNA damage response profile that was very prominent. That actually makes a lot of sense. If you remember, I told you that, when telomeres become short, they will become prone to fusions, and now when they fuse, then chromosomes will get ripped apart as they try to go through the ensuing cell divisions, because the chromosomes with fused telomeres will now have two centromeres that will pull apart in mitosis, breaking the chromosomes at random positions, and causing essentially genomic havoc, which can quickly lead to cell death if the cells try to keep dividing.
And there’s a damage response that takes place when DNA breaks appear, and this is what we see, so this makes very good sense. At senescence, we see a damage response. What was very intriguing was that we also saw others things. We saw what’s called an “environmental stress” cellular response. This is a common set of genes that are upregulated and downregulated in response to various insults, chemical insults of various kinds, but it’s a common set of genes, so this is called the environmental stress response, and we saw that response occurring, particularly at senescence. And there was a change to an aerobic metabolism program, and indeed we found that the number of mitochondria at senescence went up enormously. Very intriguing observations, not expected. Now, the survivors were fascinating because, even though I showed you they appear to grow quite well, they had a gene expression transcriptional profile which was distinct from the wild-type cells. And what persisted was a subset of the environmental stress response genes that stayed on, so what’s interesting is that those cells, which appear to be growing well on the surface, “stoics,” they’re really hurting; their physiology is different, and they sense themselves as being under what they sense as a cellular stress.
So, you can have cells that grow well without telomerase, but if you look at the cells, these cells are behaving as though under an environmental stress, and so they indicate by their gene expression profiles a cellular stress response. So in fact, cells do grow better with telomerase than without it, even though superficially they look similar, cells growing without telomerase maintaining their telomeres via this recombination type of mechanism, which is presumably somewhat more haphazard, is putting a continuous stress on the cells, even though they’ve adapted to it by a stress response. So, what we learned then is that yeast lacking telomerase, using recombination to maintain telomeres, do grow well, but they’re under continuing cellular stress, and this is what these gene expression profile analyses showed us. So, let’s go back to telomerase and consider again what it’s doing. If you have plenty of telomerase, as indeed yeast cells normally do, unless we genetically or in other ways ablate the action of telomerase… if you have plenty of telomerase, then there’s kind of a homeostasis of the telomeres.
The telomeres stay within a certain range of lengths, and remember I said at the very beginning of this lecture that telomeric repeats are different in different molecules of a population of cells, in different DNA molecules. And in fact, they distribute around an upper and a lower limit. And telomerase is one of the things that’s crucial for keeping them within this limit, and if you don’t have telomerase, then because of the DNA replication problems, they gradually fall below. But they don’t get too long either, and a great many factors limit the action of telomerase on telomeres as well, so they don’t get too long, and telomerase is also regulated on telomeric ends, so that as the telomeres get shorter, then telomerase has a higher probability of acting on the telomeres.
This is a complex system, we call it a homeostasis type of system, and it balances out the lengthening and shortening processes, so that the net result is that the telomeres stay some average length within limits, and therefore the cells keep dividing. Now, everything I’ve said implied that, if you took away telomerase, there would be quite a delay before any response was seen, and indeed, at a gross level that is true. But if one scrutinizes the cells very carefully, one sees that actually, things are a little bit different. So an experiment in yeast was done, which was to remove telomerase, as I showed you, and if you look at the cells, I showed you growing on the plate, lots of colonies, right away things look pretty okay. But if one used very sensitive, quantitative molecular probes to look at the telomeres in those cells, still with their long telomeres, one found that there were occasional… every few thousand cells or so had very short telomeres, and they would actually fuse with a DNA break that was induced to occur in those cells, something a telomere never should do if it’s a properly kept, functional telomere.
So, one in a few thousand cells had dysfunctional telomeres, as manifested by the fact that they underwent these normally forbidden fusion events. So immediately, there was a response, even if one made the telomeres all longer and then took away telomerase, one still saw this, even when the bulk telomeres were long, one could see this kind of aberrant dysfunctional telomeres immediately after loss of telomerase. So this is one piece of evidence that telomerase actually is protecting the ends of telomeres, even when they’re plenty long enough, telomerase itself seems to protect and sit at the ends of telomeres, is one interpretation, and protects them from catastrophic shortening and fusioning to a double-strand break, even when telomeres are long. This doesn’t happen in a high number of the cells, but it happens measurably, and so this protective function of telomerase is important. So I’ve introduced you to telomerase, I’ve shown you how it’s important for the long-term growth of cells because it is necessary for continuous replenishment of telomeres, and I’ve introduced you to the first piece of experimental evidence that telomerase has a protective function in cells, even when telomeres are long.
So in the next lecture, we’ll talk about telomerase and more of its protective functions in different cellular settings..