Optogenetics is an emerging and powerful technique that allows to control protein activity with light. Using optogenetics for example we can control the intracellular localisation of a protein, protein-protein interactions, or in case of enzymes their catalytic activity. Optogenetics was developed by a group of neuroscientists when they used this technique to control neuronal activity with light. But it’s only recently thanks to new photosensitive modules that optogenetics can be used in living organisms to control cell activity, for example, during embryonic development. In our lab at EMBL in Heidelberg, we use optogenetics and in particular the CRY2 / CIB1 system to control protein activity in living Drosophila developing embryos. Basic research on plant development has given rise to new tools to manipulate various processes in non-neuronal biology In 2010, Chandra Tucker and colleagues engineered the photosensitive protein domain of CRY2 and an N-terminal truncated version of CIB1 called CIBN, in such a way that they can be used as protein tags to control a target protein’s function in living biological systems.
Up until now, the CRY2 / CIB1 dimerisation system has successfully been applied to optically control numerous biological processes, such as membrane composition, transcription, receptor signalling and cytoskeletal dynamics. We were always looking for a way to control embryonic development in a very precise manner. However, with the techniques that we use so far such as for example knockout, knockdown or temperature sensitive alleles, we could never achieve the desired spatial and temporal precision. With optogenetics now, we can control embryonic development with the precision of a pulse of laser light and therefore look at very immediate cell and tissue responses upon this perturbation. Using optogenetics we can address very general questions. For example in our lab we use the CRY2 / CIB1 system to control the plasma membrane recruitment of an enzyme that allows us to modulate lipid composition and therefore achieve control over actin polymerisation during embryonic development.
Using this technique we can now for example specifically modulate cell contractility during morphogenesis of the Drosophila embryo, and ask what is the contribution of the particular cell that we are targeting to the given morphogenetic processes that we are interested in characterising. We can also use this technique to look at the response of the non-activated tissue to such perturbations. We are now going to explain the major steps of how you can implement optogenetics, and we will show you how we perform an optogenetic experiment on the early fly embryo. We start by using the Gibson assembly cloning technique to clone the protein fusion constructs into regular UAS vectors that allow for GAL4-inducible expression of the optogenetic fusion proteins in the desired cells or tissues – in our case the early Drosophila embryo. In order to start transgene expression, transgenic flies are generated and crossed to specific driver lines that provide the GAL4 transcription factor. In order to harvest the fly eggs,we put the flies in a cage with a removable agar plate supplemented with apple juice and yeast at the bottom. It is essential to keep flies that express light-sensitive fusion proteins in the dark to avoid side effects and premature light activation.
After four hours we remove the plate and prepare the embryos for microscopy. At this stage is particularly important to prevent premature light activation of the specimens. However, as the CRY2 / CIB1 system is only sensitive to blue light we don’t have to work in complete darkness and can instead work safely in red light. Halocarbon oil is added onto the plate and embryos of the desired stage are selected. Next, the non-transparent eggshell of the fly embryo – the so called chorion – is chemically removed using a chemical bleach. Finally dechorinated embryos are positioned and aligned on a Matek glass button dish and the sample is placed on the stage of the microscope To summarise the main steps of sample preparation: You start by cloning your ontogenetic constructs. You then use those to generate transgenic fly lines. Then you drive the expression of the optogenetic components in your desired tissue, which in our case is the Drosophila embryo.
And then you mount your samples in the dark. Having prepared the sample, we now need to prepare the microscope. We can efficiently activate CRY2 using two-photon illumination of light with a wavelength of 950 nanometers. Only two-photon microscopy enables us to achieve localised activation pattern with cellular resolution. Due to light scattering and due to the high photosensitivity of CRY2, conventional confocal microscopy with single photons would result in a broad and global activation of the optogenetic system. In our lab we use a commercial confocal microscope equipped with a multi-photon pulsed laser.
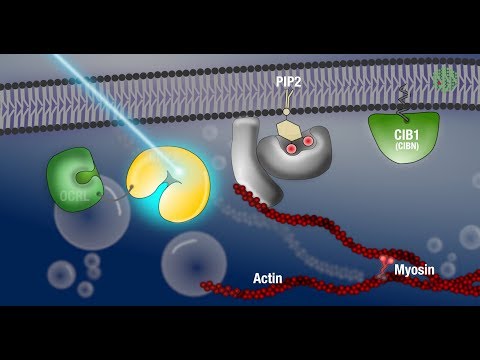
To locate, position and orientate the sample under the microscope any non blue light can be used. To monitor your protein of interest without activating the optogenetic system, you will need to use a red fluorescent protein tag such as mCherry, which can be excited using non blue light. In the optogenetic system that has been developed in our lab, CIBN is anchored at the plasma membrane and CRY2 is fused to the catalytic domain of a phosphatase called OCRL. In the dark – meaning in the absence of blue light illumination – OCRL-CRY2 are cytoplasmic and the embryo develops normally.
If you are the CRY2 / CIBN system or any other optogenetic technique, it is essential to define the dark state activity of your system and to control for potential effects in the absence of light. In our lab, we’re interested in the process of ventral furrow formation, which is a well-established model system for tissue invagination. During this process the cells in the middle axis of the embryo start to change the geometry of their upper surface and their shape. This results in the inward folding of the tissue, giving rise to the mesodermal germ layer.
If we now want to interfere with this morphogenetic event by manipulating the membrane composition through recruitment of OCRL in a specially restricted area of the embryo, we need to specify the region of activation and the size of the tissue that we want to activate. As we only want to interfere with the apical membrane, we set an activation stack of five micrometers. Since the extent of CRY2 activation depends on the laser power, we also specify the optimal laser power based on knowledge from previous experiments We use a macro to alternately image the mCherry-tagged CRY2-OCRL in the whole embryo using red light and to specifically activate the region of interest using a 950 nanometer multiphoton laser. The light induced interaction of CRY2 and CIB1 triggers protein translocation within milliseconds, and is reversible with a half life time about eight minutes. Depending on your own experimental scheme you may need to maintain or repeat the light activation. Embryos in the dark or imaged with red light form normal ventral furrow.
When we now start the microscope and shine blue light on the embryo OCRL-CRY2 gets activated and CRY2 gets recruited to CIB1at the membrane. Once at the membrane, OCRL starts a signalling cascade that leads to the depolymerisation of actin at the cortex and eventually blocks contractility of the cells. With the specificity we can achieve with optogenetics, it is even possible to activate the embryo in different pattern. This allows us to study the function of different groups of cells in the tissue and their contribution to morphogenesis. Our experimental results suggest that this contractility is essential for driving these cell shape changes that lead to the invagination of cells and folding of the embryonic tissue.
The light-induced recruitment of OCRL-CRY2 blocks contractility and folding of the embryonic tissue in the activated area of the embryo, with strong impact on neighboring cells and the embryo as a whole. The capabilities of controlling protein activity in living organisms with the precision of a laser light, essentially with cellular precision and in the second range, is opening new frontiers for the investigation of developmental pathways controlling animal morphogenesis. Now that we have this technique in our hands, we are very excited to share it with other labs that want to shed light on the mechanism of embryonic development. .
As found on Youtube